Utopedia Entry | Flagship Knowledge Article | THE TLR4/NF-κB PATHWAY
Introduction
Imagine you’re home alone late at night. You hear a strange noise downstairs. Your heart races. Your body floods with adrenaline. You’re alert, focused, ready for anything.
This is your body’s emergency response—perfectly designed to protect you from threats.
Now imagine that noise never stops. The adrenaline never fades. Your heart never stops racing. You lie awake night after night, your body screaming DANGER even when there’s nothing there. Eventually, you crash. You can’t sleep. You can’t think. You can’t function.
This is what happens inside the brain when one specific pathway—the TLR4/NF-κB pathway—gets stuck in the “on” position.
It’s the brain’s emergency broadcast system. And when it malfunctions, it doesn’t just broadcast warnings. It broadcasts destruction.
This is the story of that pathway—how it protects us, how it turns against us, and what scientists are learning about how to turn down the volume without silencing the alarm completely.
Meet the Players
What Is a Pathway Anyway?
Before we dive into the complicated names, let’s get clear on what we’re talking about.
A signaling pathway is like a game of telephone. One molecule passes a message to the next, which passes it to the next, until finally something happens. In the case of TLR4/NF-κB, that “something” is inflammation.
Think of it this way:
| Step | What Happens | Analogy |
|---|---|---|
| 1 | Something triggers the first molecule | Someone knocks on your door |
| 2 | The first molecule activates the second | You tell your spouse someone’s at the door |
| 3 | The second activates the third | Your spouse tells your kid to get the door |
| 4 | The chain reaches the nucleus | The kid opens the door |
| 5 | Genes turn on, inflammation begins | You greet your visitor |
That’s all a pathway is—a chain of molecular events that carries a signal from the outside of a cell to its command center.
The Two Main Characters
Our story has two starring molecules. Their names are complicated, but their roles are simple.
TLR4 stands for Toll-like receptor 4. “Toll-like” is a weird name—it comes from a German word “Toll” meaning “amazing” or “crazy,” because when scientists first discovered these receptors in fruit flies, they thought the flies looked amazing/crazy. The name stuck.
What matters is what TLR4 does: it sits on the surface of certain cells (especially microglia, the brain’s immune cells) and listens for danger.
NF-κB stands for nuclear factor kappa-light-chain-enhancer of activated B cells. That’s a mouthful. Just remember it as the master switch. When NF-κB gets activated, it travels to the cell’s nucleus and flips on dozens of inflammatory genes at once.
TLR4 is the ear. NF-κB is the megaphone.
Together, they form the brain’s most powerful early warning system.
DID YOU KNOW?
The Toll receptor was first discovered in fruit flies in 1985. Scientists weren’t studying inflammation—they were studying how fly embryos develop. It took nearly 10 more years before anyone realized that similar receptors in humans control immune responses. Evolution reused a good idea: the same basic system that helps a fly develop also helps your brain detect danger.
What Triggers the Alarm?
The Good Guys: Real Threats
TLR4 is designed to detect specific danger signals. Think of it as a lock, and only certain keys can open it.
The most famous key is LPS (lipopolysaccharide). LPS is a molecule found on the surface of certain bacteria, especially the kind that cause serious infections. When your immune system detects LPS, it knows bacteria are present and launches a full-scale attack.
This is why scientists often use LPS in experiments. If they inject LPS into an animal, they can study inflammation on demand. It’s like pulling the fire alarm to see how the firefighters respond.
But bacteria aren’t the only thing TLR4 detects. It also responds to damage. When cells are injured or dying, they release molecules called DAMPs (damage-associated molecular patterns). These are like the “I’m hurt!” signals that injured cells send out.
Common DAMPs include:
- HMGB1 (high-mobility group box 1)—a protein that normally lives inside the nucleus but spills out when cells die
- Heat shock proteins—released by stressed cells
- Fibrinogen—a blood protein that appears when the blood-brain barrier leaks
- S100 proteins—released by damaged brain cells
So TLR4 responds to two kinds of threats:
- Invaders from outside (bacteria, viruses, fungi)
- Trouble from inside (injured or dying cells)
The Bad Guys: Misfired Alarms
Here’s where things get complicated for brain health.
In Alzheimer’s disease, amyloid-beta (the protein that forms plaques) can directly trigger TLR4. The brain sees these protein clumps as damage signals and activates the inflammatory response. But the plaques don’t go away. The alarm keeps ringing.
In Parkinson’s disease, alpha-synuclein (the protein that forms Lewy bodies) does the same thing.
In traumatic brain injury, damaged cells release DAMPs that trigger TLR4. This starts an inflammatory response meant to clean up debris. But sometimes that response continues long after the debris is cleared.
In each case, TLR4 gets stuck in the “on” position. The emergency broadcast system keeps broadcasting, even though the emergency is over—or never really ends.
DID YOU KNOW?
Your body has 10 different Toll-like receptors (TLR1 through TLR10). Each one detects different threats. TLR4 detects bacterial LPS and certain damage signals. TLR3 detects viral RNA. TLR9 detects bacterial DNA. Together, they form a comprehensive threat detection network.
Let’s walk through exactly what happens when TLR4 gets triggered. We’ll keep it simple
The Mechanism of Action
(Let me give you the full mechanical breakdown—exactly how this pathway works at the molecular level, from the moment of trigger to the final inflammatory response. No fluff, no shortcuts).
Imagine a microglial cell in your brain. On its surface sit thousands of TLR4 molecules, each like a tiny satellite dish pointed outward, constantly listening.
Something triggers one of those dishes. Maybe it’s bacterial LPS from an infection. Maybe it’s amyloid-beta from a forming plaque. Maybe it’s HMGB1 from a dying neuron.
The moment the trigger binds to TLR4, the receptor changes shape. It’s like a key turning in a lock. The door to the cell’s interior cracks open.
The Receptor: TLR4 Up Close
Let’s start with the star of the show: TLR4 itself.
Imagine TLR4 as a protein that threads through the cell membrane like a sewing needle poking through fabric. Part of it sticks outside the cell (the extracellular domain). Part of it sits inside the cell (the intracellular domain). And a middle section anchors it in the membrane.
The extracellular domain is shaped like a horseshoe. Along this horseshoe are specialized regions called leucine-rich repeats—think of them as the ridges on a key. Different arrangements of these ridges recognize different threats.
When the right trigger comes along—say, a piece of bacterial LPS or a fragment of amyloid-beta—it fits into this horseshoe like a key into a lock. This binding changes the shape of the entire receptor.
Here’s the critical part: TLR4 doesn’t work alone. When activated, it grabs hold of another protein called MD-2 (myeloid differentiation factor 2). MD-2 is like a co-pilot—it helps TLR4 recognize certain triggers, especially LPS. Together, TLR4 and MD-2 form a complex that now looks completely different than it did a moment ago.
This shape change is the signal. It tells the inside of the cell: “Something has bound to me. Prepare for action.”
The TIR Domain: The Intracellular Messenger
Now look inside the cell. The part of TLR4 that pokes into the cytoplasm contains a region called the TIR domain (Toll/IL-1 receptor domain). This is the business end of the receptor—the part that starts the internal signaling cascade.
When TLR4 changes shape on the outside, the TIR domain changes shape on the inside. It’s like squeezing one end of a balloon—the other end bulges out.
This bulging creates a docking station. Proteins inside the cell can now grab onto the TIR domain. The first proteins to arrive are the adapters.
DID YOU KNOW?
The TIR domain is named after the IL-1 receptor because scientists discovered that Toll-like receptors and interleukin-1 receptors share this same signaling module. Evolution reused a successful design: the same basic structure handles both infection signals (through TLRs) and injury signals (through IL-1 receptors).
The Adapters: MyD88 and TRIF
Two main adapter proteins carry the signal forward. Think of them as lieutenants taking orders from the general (TLR4).
MyD88 (myeloid differentiation primary response 88) handles the rapid response. Within seconds of TLR4 activation, MyD88 binds to the TIR domain. It then recruits a family of kinases called IRAKs (IL-1 receptor-associated kinases).
The sequence goes:
- MyD88 brings in IRAK4
- IRAK4 activates IRAK1 by adding phosphate groups
- Activated IRAK1 then recruits TRAF6 (TNF receptor-associated factor 6)
TRIF (TIR-domain-containing adapter-inducing interferon-β) handles a slower, longer-lasting response. Instead of binding directly to TLR4, TRIF binds through another adapter called TRAM (TRIF-related adapter molecule). TRIF then activates a different pathway involving TBK1 and IKKε, leading to production of interferons—antiviral molecules.
Why two pathways? Because different threats require different responses. Bacteria (detected by TLR4) might need a strong inflammatory response. Viruses (detected by other TLRs) might need more interferon production. The cell tailors its response to the threat.
For neuroinflammation, the MyD88 pathway is the main player. That’s where we’ll focus.
TRAF6: The Signal Amplifier
TRAF6 is a fascinating molecule. It’s an E3 ubiquitin ligase—an enzyme that tags other proteins with a small molecule called ubiquitin.
Ubiquitin is best known for marking proteins for destruction (like putting a “trash” label on them). But TRAF6 uses a special type of ubiquitin linkage called K63-linked polyubiquitin that doesn’t send proteins to the trash. Instead, it creates a scaffold—a platform where other signaling molecules can gather.
Think of TRAF6 as building a molecular campfire. It brings in wood (ubiquitin chains) and then invites other molecules to sit around the fire and talk.
The molecules that gather include TAK1 (TGF-beta-activated kinase 1) and its binding partners TAB1, TAB2, and TAB3. TAK1 is a kinase—an enzyme that phosphorylates (adds phosphate to) other proteins. When TAK1 sits on the ubiquitin scaffold created by TRAF6, it becomes activated.
Now we have a fully operational signaling complex. The message has been passed from TLR4 → MyD88 → IRAK4 → IRAK1 → TRAF6 → TAK1. Each step has amplified the signal. One TLR4 activation leads to hundreds of TAK1 molecules working.
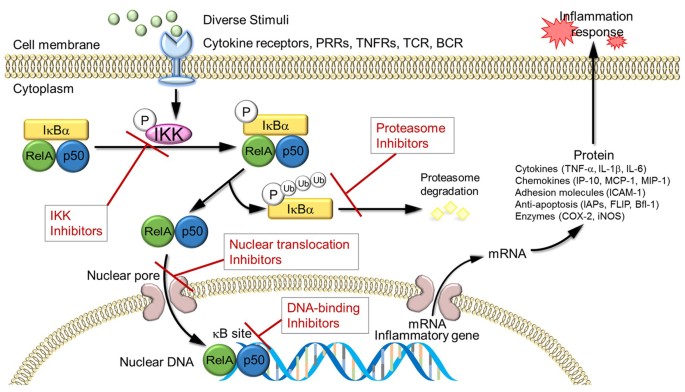
The IKK Complex: The Gatekeeper
TAK1 now activates the next major player: the IKK complex (IκB kinase complex).
The IKK complex is made of three parts:
- IKKα (IKK-alpha)
- IKKβ (IKK-beta)
- NEMO (NF-κB essential modulator), also called IKKγ
Think of IKKα and IKKβ as the workers and NEMO as the manager. NEMO organizes the complex and ensures it goes where needed.
TAK1 phosphorylates IKKβ, activating it. Now the IKK complex is ready for its job.
And what is that job? It’s time to release NF-κB from its cage.
The Cage: IκB and NF-κB
Remember NF-κB, our master switch? It normally sits in the cytoplasm, held captive by a protein called IκB (inhibitor of κB).
IκB physically covers up the part of NF-κB that says “go to the nucleus.” Think of IκB as a blindfold and handcuffs. NF-κB can’t see where to go (the nucleus) and can’t move even if it could see.
There are several forms of IκB (IκBα, IκBβ, IκBε), but IκBα is the most important for rapid responses.
The IKK complex’s job is to remove these handcuffs. IKK phosphorylates IκB at two specific spots. This phosphorylation is like painting a target on IκB.
Once phosphorylated, IκB is recognized by another protein complex called SCF-βTrCP. This complex adds a different type of ubiquitin—the kind that does send proteins to the trash. K48-linked polyubiquitin chains are added to IκB, marking it for destruction.
The proteasome—the cell’s garbage disposal—now destroys IκB. It chews it up into tiny pieces.
NF-κB is free.
DID YOU KNOW?
The proteasome is a massive protein complex shaped like a barrel with caps on both ends. Proteins tagged for destruction are fed into one end, chopped into small peptides inside the barrel, and spit out the other end. This system clears out damaged or unwanted proteins—including our friend IκB once it’s been marked.
Nuclear Entry: The Master Switch Flips
Free NF-κB now reveals its nuclear localization signal—a molecular “address label” that says “deliver to nucleus.” This signal was always there, but IκB was covering it up.
The cell has a transport system for moving things into the nucleus. Proteins called importins grab onto NF-κB and guide it through nuclear pores—gateways in the nuclear membrane.
Once inside the nucleus, NF-κB searches for specific DNA sequences called κB sites. These are short stretches of DNA with a particular pattern: GGGRNNYYCC (where R is any purine, N is any nucleotide, and Y is any pyrimidine).
Think of κB sites as docking stations. NF-κB binds to them and then recruits other proteins called coactivators that help unwind DNA and attract the machinery that reads genes.
Now the genes turn on.
The Genetic Program: What Gets Turned On
NF-κB doesn’t activate just one gene. It activates hundreds. This is why it’s called a master switch—it controls an entire program of inflammation.
The genes fall into categories:
Cytokines and chemokines (signaling molecules):
- TNF-α (tumor necrosis factor-alpha)—activates other immune cells, promotes inflammation
- IL-1β (interleukin-1 beta)—fever, inflammation, activation of more immune cells
- IL-6 (interleukin-6)—promotes inflammation, acute phase response
- IL-8 (interleukin-8)—attracts neutrophils to the site
- MCP-1 (monocyte chemoattractant protein-1)—attracts monocytes/macrophages
Enzymes that produce inflammatory mediators:
- COX-2 (cyclooxygenase-2)—produces prostaglandins (pain, fever, inflammation)
- iNOS (inducible nitric oxide synthase)—produces nitric oxide (kills microbes, but also damages tissue)
- PLA2 (phospholipase A2)—produces precursors for inflammatory lipids
Adhesion molecules (help immune cells stick to blood vessels and enter tissue):
- ICAM-1 (intercellular adhesion molecule 1)
- VCAM-1 (vascular cell adhesion molecule 1)
- E-selectin
Immune receptors and activators:
- More TLRs (amplifying the response)
- MHC molecules (present antigens to T-cells)
- Co-stimulatory molecules (activate T-cells)
Anti-apoptotic factors (help inflammatory cells survive):
- Bcl-2 family members
- c-IAPs (inhibitor of apoptosis proteins)
Notice that last category? NF-κB actually helps inflammatory cells survive longer. This is great for fighting infections—you want those soldiers to live. But in chronic inflammation, it means the inflammatory cells stick around longer than they should.
The Feedback Loops: Why It Gets Stuck
Now here’s the really important part for understanding chronic neuroinflammation: NF-κB also turns on its own inhibitors.
One of the genes NF-κB activates is IκBα—the very protein that cages NF-κB. This creates a negative feedback loop:
- NF-κB enters nucleus
- It turns on inflammatory genes
- It also turns on IκBα gene
- New IκBα protein is made
- IκBα enters nucleus, grabs NF-κB, and drags it back to the cytoplasm
- Inflammation stops
This is how the system normally resets. The fire burns, then the sprinklers turn on.
But in chronic inflammation, something breaks this loop. The triggers persist (amyloid plaques, ongoing damage), so TLR4 keeps getting activated. The signal keeps coming. The IκBα that’s made gets quickly destroyed by continuous IKK activity.
The system can’t reset because the alarm won’t stop ringing.
The Amplification Loop: Cytokines Activate More TLR4
Here’s where things spiral.
TNF-α and IL-1β (produced by NF-κB activation) don’t just affect other cells. They can also bind to receptors on the same cell that produced them, or on neighboring cells.
TNF-α binds to TNFR1 (TNF receptor 1). IL-1β binds to IL-1R (IL-1 receptor). Both of these receptors activate—you guessed it—NF-κB.
So now you have a positive feedback loop:
- TLR4 activates NF-κB
- NF-κB produces TNF-α and IL-1β
- TNF-α and IL-1β activate more NF-κB through their own receptors
- Even more cytokines are produced
- The inflammation spreads to neighboring cells
This is how a local inflammatory response becomes a global one. Each activated cell becomes a broadcasting station, sending signals that activate its neighbors.
DID YOU KNOW?
This cytokine amplification loop is why anti-TNF drugs (like Humira) work for inflammatory diseases. By blocking TNF-α, they interrupt this self-amplifying cycle. These drugs are used for rheumatoid arthritis and Crohn’s disease, and researchers are exploring whether they might help certain brain conditions.
The Synergy with Other Pathways
TLR4/NF-κB doesn’t operate in isolation. It talks constantly with other inflammatory pathways:
NLRP3 inflammasome: NF-κB turns on the genes for pro-IL-1β and NLRP3. Then a second signal (like ATP or amyloid crystals) activates the inflammasome, which processes pro-IL-1β into active IL-1β. TLR4 provides the first hit; NLRP3 provides the second. Together, they produce massive IL-1β.
MAPK pathways: TLR4 also activates p38, JNK, and ERK pathways through TRAF6 and TAK1. These pathways produce their own inflammatory mediators and can enhance NF-κB activity.
PI3K/Akt pathway: This cell survival pathway can be activated downstream of TLR4, helping inflammatory cells survive longer.
JAK-STAT pathway: Cytokines produced by NF-κB (like IL-6) activate JAK-STAT signaling, which produces more inflammatory mediators.
These pathways form a network. Block one, and the others can sometimes compensate. This is why single-target drugs often fail—the system is too interconnected.
Visual Summary: The Complete TLR4/NF-κB Cascade
text
1. TRIGGER BINDS
↓
LPS, amyloid-beta, DAMPs bind to TLR4 + MD-2
↓
2. RECEPTOR ACTIVATION
↓
TLR4 changes shape → TIR domain exposed
↓
3. ADAPTER RECRUITMENT
↓
MyD88 binds → recruits IRAK4 → activates IRAK1
↓
4. SIGNAL AMPLIFICATION
↓
IRAK1 recruits TRAF6 → TRAF6 builds ubiquitin scaffold
↓
5. KINASE ACTIVATION
↓
TAK1 binds scaffold → becomes active → activates IKK complex
↓
6. CAGE DESTRUCTION
↓
IKK phosphorylates IκB → IκB ubiquitinated → IκB destroyed by proteasome
↓
7. NF-κB RELEASE
↓
NF-κB free → enters nucleus → binds DNA
↓
8. GENE ACTIVATION
↓
Inflammatory genes turn on: TNF-α, IL-1β, IL-6, COX-2, iNOS, etc.
↓
9. FEEDBACK LOOPS
↓
IκBα produced (tries to stop) + cytokines produced (amplify signal)
↓
10. CHRONIC STATE
↓
Persistent triggers → persistent activation → chronic neuroinflammation
The Takeaway: Why Mechanism Matters
Understanding this molecular dance isn’t just academic. It reveals specific points where we might intervene:
| Step | Target | Approach |
|---|---|---|
| 1 | TLR4 | Block receptor with antagonists |
| 2 | MyD88 | Inhibit adapter recruitment |
| 3 | IRAK4 | Kinase inhibitors |
| 4 | TRAF6 | Disrupt ubiquitin scaffold |
| 5 | TAK1 | Kinase inhibitors |
| 6 | IKK | Kinase inhibitors (like BMS-345541) |
| 7 | NF-κB nuclear entry | Block nuclear transport |
| 8 | DNA binding | Block NF-κB-DNA interaction |
| 9 | Cytokine receptors | Block TNF or IL-1 receptors |
| 10 | Multiple pathways | Combination therapy |
Each intervention point has its own advantages and challenges. Blocking TLR4 is very specific but might leave other inflammatory pathways untouched. Blocking NF-κB directly is powerful but could block protective functions too.
The future lies in understanding which intervention works best for which disease, at which stage, in which patient.
Now things get really interesting. The products NF-κB creates don’t just affect the original cell. They spread through the surrounding tissue, affecting other cells.
TNF-α from one microglial cell activates TLR4 on neighboring microglia. They start their own NF-κB cascades. The inflammation spreads like a wave.
Cytokines also signal to astrocytes, pulling them into the inflammatory response. They change shape, release their own signals, and the inflammation amplifies.
This is how a tiny trigger—a few amyloid plaques, a minor injury—can eventually lead to brain-wide inflammation. The signal propagates, cell to cell, through the TLR4/NF-κB pathway.
DID YOU KNOW?
The entire TLR4/NF-κB cascade—from trigger to gene activation—happens in minutes. Your cells respond to threats faster than you can read this sentence. That speed is essential for survival, but it also means inflammation can spiral out of control quickly.
Why This Matters for Brain Health
The Good: Protection
Let’s not demonize this pathway. Without TLR4/NF-κB, you’d be dead.
When bacteria enter the brain, this system detects them and launches an attack. When neurons are injured, it clears the debris and promotes healing. When viruses infect brain cells, it helps eliminate them.
The pathway is essential. It’s saved your life more times than you’ll ever know.
The Bad: Chronic Activation
The problem isn’t the pathway. It’s when the pathway won’t turn off.
In neurodegenerative diseases, triggers persist. Amyloid plaques don’t disappear. Tau tangles don’t dissolve. Damaged cells keep releasing DAMPs. The alarm keeps ringing.
Chronic TLR4/NF-κB activation leads to:
Synapse damage: The cytokines produced by NF-κB directly harm synapses—the connections between neurons. They weaken signals, disrupt communication, and eventually cause synapses to disappear.
Neuronal death: Prolonged exposure to inflammatory cytokines pushes neurons toward apoptosis—programmed cell death. They commit suicide to escape the toxic environment.
Blood-brain barrier breakdown: Cytokines signal endothelial cells to loosen their tight junctions. The barrier becomes leaky. More inflammatory cells enter from the blood. The fire gets more fuel.
Microglial exhaustion: Microglia can’t maintain their protective functions while constantly producing inflammatory signals. They become dysfunctional, losing their ability to clear debris and support neurons.
Amplification of protein aggregation: Inflammation actually promotes more amyloid and tau accumulation. It’s a vicious cycle: aggregates trigger inflammation, inflammation promotes more aggregates.
The Ugly: Spreading to Healthy Tissue
One of the most dangerous aspects of chronic TLR4/NF-κB activation is that it doesn’t stay contained.
In Alzheimer’s, inflammation starts near amyloid plaques. But cytokines diffuse through the tissue, activating microglia far from any plaque. Healthy regions get caught in the crossfire.
In traumatic brain injury, inflammation starts at the impact site. But within days, it spreads throughout the brain. A focal injury becomes a global problem.
The TLR4/NF-κB pathway is the engine driving this spread. Each activated cell becomes a new broadcasting station, sending inflammatory signals to its neighbors.
DID YOU KNOW?
PET scans can now visualize activated microglia in living human brains using tracers that bind to TSPO (translocator protein), which increases on activated microglia. These scans show that neuroinflammation spreads over time—and the pattern of spread predicts cognitive decline.
The Pathway in Specific Diseases
Alzheimer’s Disease
In Alzheimer’s, amyloid-beta directly triggers TLR4 on microglia. This starts an inflammatory response meant to clear the plaques. But the plaques persist, so the inflammation persists.
Genetic studies confirm the importance of this pathway:
- TREM2 (a receptor that normally dampens inflammation) variants increase Alzheimer’s risk
- CD33 (another immune regulator) variants also affect risk
- APOE4 (the strongest genetic risk factor) impairs microglial function
Each of these genes connects back to TLR4/NF-κB signaling. TREM2 normally inhibits TLR4. When TREM2 is broken, TLR4 signaling goes unchecked. CD33 regulates microglial activation. APOE4 affects how microglia respond to damage.
The pathway sits at the center of Alzheimer’s biology, linking protein aggregation to neurodegeneration.
Parkinson’s Disease
In Parkinson’s, the trigger is alpha-synuclein. When neurons die (for whatever reason), they release alpha-synuclein into the surrounding tissue. Microglia detect it through TLR4 and become activated.
The resulting inflammation kills more neurons, releasing more alpha-synuclein. Another vicious cycle.
PET imaging studies show that microglial activation in Parkinson’s correlates with symptom severity and progresses as the disease advances.
Multiple Sclerosis
MS is an autoimmune disease, but TLR4/NF-κB still plays a central role.
Autoreactive T-cells enter the brain and attack myelin. This triggers massive cell death, releasing DAMPs that activate TLR4 on microglia and astrocytes. The resulting inflammation amplifies the damage and recruits more immune cells.
In progressive MS, this smoldering inflammation continues even without new T-cell attacks. The pathway has become self-sustaining.
Traumatic Brain Injury and Stroke
Acute injuries trigger immediate TLR4 activation through DAMPs released by dying cells. This acute inflammation helps clear debris and promote repair—if it resolves properly.
But in many cases, it doesn’t. Studies show microglial activation can persist for years after a single TBI. These individuals have chronically elevated inflammatory markers and increased risk of later dementia.
Depression and Other Psychiatric Conditions
Even conditions not traditionally considered “inflammatory” involve TLR4/NF-κB.
Chronic stress activates this pathway. Stress hormones can directly trigger inflammatory signaling, and stressed neurons release DAMPs. Patients with major depression have elevated inflammatory markers, and anti-inflammatory treatments can improve symptoms in some cases.
The mind-inflammation connection is real, and TLR4/NF-κB is part of it.
Turning Down the Volume
The Challenge: Don’t Silence, Modulate
If TLR4/NF-κB is essential for survival, you can’t just block it completely. That would leave the brain defenseless against real threats.
The goal is modulation—turning down the volume without silencing the alarm entirely.
Think of it like a smoke detector. You want it to go off when there’s a real fire. You don’t want it to go off every time you burn toast. And you definitely don’t want it stuck on “alarm” for years.
Natural Compounds That Modulate the Pathway
Nature has provided many compounds that can gently influence TLR4/NF-κB signaling:
Curcumin (from turmeric) inhibits NF-κB activation. It’s been used in traditional medicine for centuries, and modern research confirms its anti-inflammatory properties. The challenge is getting enough into the brain—curcumin is poorly absorbed.
Resveratrol (from grapes and red wine) also inhibits NF-κB. It activates sirtuins (longevity proteins) that dampen inflammatory signaling.
EGCG (from green tea) modulates multiple inflammatory pathways, including TLR4/NF-κB.
Harmine (from a plant used in traditional medicine) inhibits both TLR4/NF-κB and the NLRP3 inflammasome (another inflammatory pathway). Recent research shows it reduces inflammation in animal models of Alzheimer’s and stroke.
Osthole (from a plant used in traditional Chinese medicine) inhibits NEU1, an enzyme that amplifies TLR4 signaling. It reduces microglial activation and improves cognition in animal models.
These compounds are not strong enough to treat advanced disease, but they may help maintain healthy inflammatory balance when used preventively.
DID YOU KNOW?
Many of these natural compounds work through something called “hormesis”—they create mild stress that strengthens the cell’s defense systems. A little bit of stress makes the system more resilient, just like exercise stresses muscles to make them stronger.
Pharmaceutical Approaches
Drug companies are developing more targeted interventions:
TLR4 antagonists block the receptor directly, preventing triggers from activating it. Several are in development for sepsis (where the pathway goes into overdrive) and could potentially be repurposed for brain diseases.
IKK inhibitors block the IKK complex, preventing it from releasing NF-κB from its cage. These are potent anti-inflammatories, but safety concerns exist—completely blocking NF-κB for long periods might have side effects.
NEU1 inhibitors target neuraminidase 1, an enzyme that amplifies TLR4 signaling. Recent research suggests this approach can reduce neuroinflammation while preserving protective functions.
cPLA2 inhibitors target calcium-dependent phospholipase A2, an enzyme linked to inflammation in APOE4 carriers. These compounds cross the blood-brain barrier and reduce neuroinflammation in preclinical models.
FGF20 (fibroblast growth factor 20) is a protein that suppresses TLR4/NF-κB while enhancing protective microglial functions. It’s early-stage research but shows promise.
The Blood-Brain Barrier Problem
Any drug targeting the brain must cross the blood-brain barrier. This is a major challenge for anti-inflammatory therapies.
Some strategies:
- Small molecules designed specifically to penetrate the barrier
- Nanoparticles that carry drugs across
- Trojan horse approaches that disguise drugs as nutrients the barrier normally transports
- Focused ultrasound to temporarily open the barrier at specific locations
Lifestyle Approaches That Modulate the Pathway
Before drugs, there’s lifestyle. Multiple studies show that everyday choices influence TLR4/NF-κB activity:
Exercise reduces TLR4 expression on immune cells and dampens NF-κB activation. Regular exercisers have lower baseline inflammation and faster resolution after injury.
Diet matters enormously. The Mediterranean diet (rich in polyphenols, omega-3s, and fiber) reduces inflammatory signaling. Omega-3 fatty acids (fish oil) can directly inhibit TLR4.
Sleep is critical. During sleep, the brain clears inflammatory debris. Sleep deprivation increases TLR4 expression and NF-κB activation.
Stress reduction works. Mindfulness meditation has been shown to reduce NF-κB activation and inflammatory gene expression. The mind truly influences the immune system.
Caloric restriction and intermittent fasting reduce inflammation through multiple mechanisms, including dampening TLR4/NF-κB signaling.
None of these are magic bullets. But together, they create an environment where the pathway is less likely to get stuck in the “on” position.
The Future—Precision Modulation
Biomarkers: Who Needs Treatment?
Not everyone with amyloid plaques develops Alzheimer’s. Not everyone with a head injury develops chronic inflammation.
We need ways to identify whose TLR4/NF-κB pathway is stuck “on”—and intervene before damage accumulates.
Promising biomarkers include:
- PET imaging of microglial activation (TSPO-PET)
- CSF inflammatory markers (sTREM2, YKL-40, cytokines)
- Blood inflammatory markers (GFAP, NFL, inflammatory cytokines)
- Genetic risk profiles (APOE, TREM2, CD33 variants)
The goal is precision medicine: treat the people who need it, spare those who don’t.
Timing: When to Intervene
The pathway may be most treatable early, before inflammation becomes self-sustaining.
In Alzheimer’s, this means intervening during the preclinical phase—when amyloid is accumulating but cognitive symptoms haven’t appeared. In TBI, this means monitoring long-term inflammation and treating those whose inflammation doesn’t resolve.
Timing matters as much as targeting.
Combination: Multiple Pathways
TLR4/NF-κB doesn’t work alone. It interacts with:
- NLRP3 inflammasome—amplifies IL-1β production
- MAPK pathways—parallel inflammatory cascades
- JAK-STAT pathway—responds to cytokines
- TREM2 pathway—attempts to dampen inflammation
Future treatments will likely target multiple pathways simultaneously, just as cancer treatments combine multiple drugs.
The Ultimate Goal: Resilience
The best strategy is building brains that don’t get stuck in chronic inflammation in the first place.
This means:
- Preventing the triggers (amyloid accumulation, TBI, infections)
- Building reserve through lifestyle (exercise, diet, sleep, stress management)
- Early detection and intervention when inflammation starts
- Personalized approaches based on genetics and biomarkers
We may never completely eliminate neuroinflammation. But we can learn to control it—to keep the emergency broadcast system ready to respond without getting stuck broadcasting forever.
The Volume Knob
The TLR4/NF-κB pathway is neither good nor bad. It’s a tool—an exquisitely sensitive early warning system that has protected our brains for millions of years.
The problem isn’t the tool. It’s that in modern life, with chronic diseases and long lifespans, the alarm gets stuck. The fire that should burn out and be extinguished keeps smoldering for decades, slowly consuming the tissue it was meant to protect.
Understanding this pathway gives us a volume knob. We can’t—and shouldn’t—turn it off completely. But we can learn to turn it down. We can learn to distinguish real fires from false alarms. We can learn to let the system rest.
The research is moving fast. New compounds, new biomarkers, new understanding emerge every year. The day may come when checking your neuroinflammation status is as routine as checking your cholesterol—and treating it as straightforward.
Until then, we have what we’ve always had: the knowledge that lifestyle matters, that inflammation is real, and that the brain’s hidden fire can be controlled.
DID YOU KNOW? (Bonus Round)
- NF-κB was discovered in 1986 by David Baltimore and colleagues. They were studying how antibodies are produced in B cells and found a protein that binds to the “κ light chain” gene enhancer. They named it “nuclear factor that binds to the κ light chain enhancer”—NF-κB. They had no idea it would turn out to control inflammation throughout the body.
- There are actually five different NF-κB proteins that combine in different ways: p50, p52, p65 (RelA), RelB, and c-Rel. Different combinations (dimers) activate different sets of genes. The most common and best-studied is the p50-p65 dimer.
- Some viruses have evolved to hijack NF-κB. HIV, for example, uses NF-κB to activate its own genes. When immune cells are activated (turning on NF-κB), they also activate the dormant virus hiding inside them.
- The same pathway that protects your brain can also cause brain cancer. In glioblastoma, NF-κB is often constitutively active, driving cancer cell survival and growth. The pathway is a double-edged sword in every tissue.
- Aspirin works partly by inhibiting NF-κB. At high doses, aspirin blocks IKK, preventing IκB degradation. This may explain some of its anti-inflammatory effects beyond COX inhibition.
This is the machinery. This is how the alarm system works. Understanding it gives us power—the power to fix it when it breaks, to turn it down when it’s too loud, and to preserve its protective functions while taming its destructive potential.
Continue Exploring…
This article is part of Utopedia’s Neuroinflammation Deep Dive Series. To deepen your understanding, explore these related entries:
Neuroinflammation: The Brain’s Hidden Fire
The flagship article introducing the full landscape of brain inflammation—now available in the Utopedia library.
Microglia: The Brain’s First Responders
Deep dive into the cells that house the TLR4/NF-κB pathway and decide when to activate it.
The NLRP3 Inflammasome: The Amplifier
How a second pathway takes TLR4’s signal and turns it into a full-blown inflammatory response.
The Blood-Brain Barrier: Fortress Under Siege
What happens when inflammation attacks the brain’s protective walls—and how TLR4 signaling contributes.
Natural Anti-Inflammatories for Brain Health
The science behind curcumin, resveratrol, omega-3s, and other compounds that modulate TLR4/NF-κB.
APOE: The Gene That Shapes Brain Aging
How the most important Alzheimer’s risk gene influences microglial function and inflammatory responses.
Clinical Trials in Neuroinflammation: 2026 Update
What’s in the pipeline targeting TLR4, NF-κB, and related pathways.
Join the UtopiaCircle Campus Network
This article is just one stop on a much larger journey. At UtopiaCircle, we’re building a community of curious minds exploring the frontiers of science, medicine, and human potential.
When you join our Campus Network, you get:
- Early access to new Utopedia entries before public release
- Monthly research summaries from leading journals
- Live Q&A sessions with researchers and clinicians
- Discussion forums to explore ideas with fellow learners
- Opportunities to suggest topics for future articles
Leave a comment below:
- Did this explanation of TLR4/NF-κB make sense?
- What other molecular pathways would you like us to simplify?
- Have you or a loved one been affected by a condition involving neuroinflammation?
Your questions shape the content we create. Your stories remind us why this work matters.
Share this article with someone who needs to understand the alarm system inside their brain.
Together, we move from curiosity to understanding.
References
- Zhu L, et al. FGF20 alleviates neuroinflammation in ischemic stroke by modulating microglial polarization via TREM2-TLR4/NF-κB pathway. Cell Immunol. 2026;423-424:105095.
- Alzarea SI, et al. A network pharmacology and in silico approach to target NEU1-mediated microglial activation in neuroinflammation: Validation in an LPS-induced mouse model. Comput Biol Med. 2025;178:109652.
- Sadybekov AV, et al. Development of potent, selective cPLA2 inhibitors for targeting neuroinflammation in Alzheimer’s disease and other neurodegenerative disorders. npj Drug Discovery. 2026.
- Shen X, et al. Harmine and its derivatives: A promising multi-target therapeutic avenue for Alzheimer’s disease. Neuroscience. 2026. Online ahead of print.
- Müller L, et al. Neuroimmune crosstalk in chronic neuroinflammation: microglial interactions and immune modulation. Front Cell Neurosci. 2025;19:1575022.
- Microglial dysfunction in Alzheimer’s disease: Mechanisms, emerging therapies, and future directions. Exp Neurol. 2025;392:115374.
- The role of inflammation in neurological disorders: a brief overview of multiple sclerosis, Alzheimer’s, and Parkinson’s disease. Front Neurol. 2024;15:1439125.
- Shi FD, Wee Yong V. Neuroinflammation Across Neurological Diseases. Science. 2025;388(6753).