Welcome to Utopedia, where we untangle systems that haunt textbooks and make them click like puzzle pieces. Today: the RAAS — a symphony of survival, a pathway both elegant and dangerous.
Imagine This…
You are a king (your body).
Your kingdom (blood circulation) needs stable water supply and pressure to keep the villages (organs) alive. One day, there’s a drought:
Who do you send?
Your loyal general, the kidneys.
You’re the king (the body). Your villages (organs) depend on steady rivers of blood. One day, there’s a drought:
- Blood pressure falls.
- Sodium levels drop.
- Volume dwindles.
Who notices first? Not the brain. Not the heart. It’s the kidneys — those unsung regulators.
Inside them, tiny guards called juxtaglomerular (JG) cells stand watch.
Step 1:The Whistleblower (Renin)
The JG cells detect crisis through three eyes:
- Baroreceptor eye → senses renal arterial pressure.
- Chemoreceptor eye (Macula densa) → monitors sodium chloride in distal tubule.
- Nerve eye → listens to sympathetic nerves releasing norepinephrine.
When all scream “low!”, they release Renin.
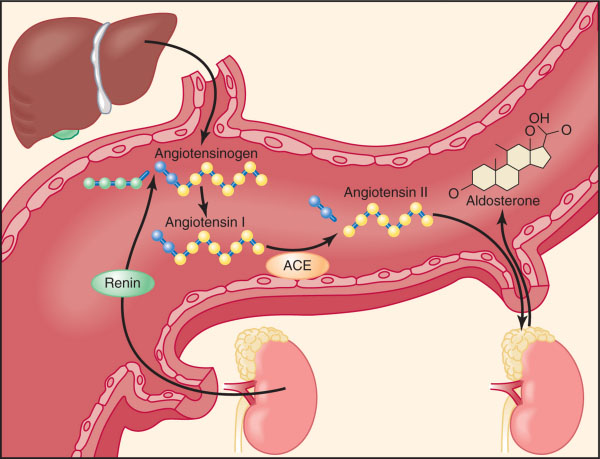
Note that Renin is not a hormone — it’s an enzyme. It cleaves angiotensinogen into action.
Step 2: The Messenger’s Transformation
Renin meets angiotensinogen, a passive liver-made plasma protein.
- Slice 1: → Angiotensin I (inactive).
- Slice 2: In the lungs, ACE sharpens Ang I into Angiotensin II.
Note: There are other enzymes too (chymase, cathepsin G) that can also generate Ang II, explaining why ACE inhibitors don’t fully block RAAS.
Step 3: Angiotensin II — The Multi-Weapon Warrior
Now we meet the real star. Ang II is small but fierce, acting on AT₁ receptors across the body:
- Blood vessels → vasoconstriction. Resistance rises, pressure jumps.
- Adrenal cortex (zona glomerulosa) → Aldosterone release.
- Kidneys → Direct sodium reabsorption in proximal tubule.
- Brain (hypothalamus) → Thirst + ADH secretion.
- Sympathetic nervous system → Enhances norepinephrine release.
Note: Ang II also acts on AT₂ receptors, which usually oppose AT₁ (vasodilation, anti-growth). This yin-yang balance is often ignored in school.
Step 4: Aldosterone — The Slow Architect
Aldosterone arrives like an engineer, working at the distal tubule and collecting duct:
- Inserts sodium channels (ENaCs) into cells.
- Boosts sodium-potassium pumps.
- Net effect: Sodium and water reabsorbed, potassium excreted.
Note: This genomic effect takes hours to days, unlike Ang II’s rapid vessel squeeze.
Step 5: The Sidekick — ADH (Vasopressin)
Although not strictly part of RAAS, ADH often joins the mission. Ang II whispers to the hypothalamus:
- Release ADH → acts on collecting ducts → inserts aquaporins → water reabsorption skyrockets.
This makes the defense water-tight (literally).
Step 6: The Feedback
Once pressure is restored, renin release should shut off.
- Negative feedback keeps balance.
- But in disease (renal artery stenosis, CHF), RAAS keeps firing — turning helpful soldiers into rebels.
When RAAS Overfires: The Villain Arc
- Hypertension → Chronic Ang II vasoconstriction.
- Heart failure → Volume overload worsens strain.
- Kidney disease → Constant sodium retention leads to edema.
- Conn’s syndrome → Tumor-driven aldosterone excess → HTN + hypokalemia.
Note: This is why RAAS blockers (ACE inhibitors, ARBs, spironolactone) are pillars of modern medicine.
Clinical Nerd Points
- ACE Inhibitors (enalapril, lisinopril): block Ang II formation → ↓ BP, ↓ afterload, ↓ remodeling.
- ARBs (losartan, valsartan): block AT₁ receptors, leaving AT₂ unopposed (vasodilatory bonus).
- Aldosterone Antagonists (spironolactone, eplerenone): potassium-sparing, lifesaving in CHF.
- Direct Renin Inhibitors (aliskiren): block the start itself (rarely used, but nerd gold).
Clinical pearl: ACE inhibitors also ↑ bradykinin (causing cough and angioedema). ARBs don’t.
The Balancing Act: RAAS vs Counterforces
Your body isn’t one-sided. RAAS raises pressure, but other systems calm it:
- Atrial Natriuretic Peptide (ANP) → Released when atria stretch, promoting sodium/water loss.
- Brain Natriuretic Peptide (BNP) → Heart failure marker, counteracts RAAS.
- Nitric Oxide → Vasodilator that opposes Ang II’s constriction.
This is the yin-yang of blood pressure control.
Why RAAS is So Confusing in School
At school, RAAS is taught as a linear flowchart:
Renin → Ang I → Ang II → Aldosterone.
But in reality, it’s a web:
- Ang II has multiple receptors with different effects.
- ACE isn’t the only enzyme making Ang II.
- RAAS interacts with ADH, SNS, ANP, and kidneys in complex loops.
This complexity is why drugs targeting different steps exist.
Interactive Recap: Fill the Map
- Which organ makes angiotensinogen?
- Which enzyme besides ACE can produce Ang II?
- Which adrenal layer makes aldosterone?
- Which receptor subtype of Ang II is pro-hypertensive?
- Which hormones directly counteract RAAS?
(Answers: Liver, chymase, zona glomerulosa, AT₁ receptor, ANP/BNP.)
Closing Utopedia Thought
RAAS is not just a system — it’s a philosophy of survival:
- Immediate (Ang II squeezes vessels).
- Intermediate (Aldosterone builds sodium stores).
- Long-term (ADH keeps water, heart remodels under stress).
Too little RAAS, and you collapse.
Too much RAAS, and you drown in your own pressure.
It’s a tightrope act between life-saving hero and disease-driving villain.
And now, you don’t just memorize it — you own it.