Utopedia Entry | Knowledge Article | Neuroinflammation
Imagine a fire burning silently inside your skull. You cannot feel it. You cannot see it. There is no smoke, no heat, no flames licking at the surface. But this fire—slow, smoldering, relentless—may be the very thing stealing memories, clouding thoughts, and eroding the person you are.
This is neuroinflammation: the brain’s own immune response gone wrong.
For decades, we believed the brain was “immune privileged”—a fortress so well-guarded that the body’s inflammatory forces simply couldn’t enter. We thought inflammation was something that happened in your joints, your skin, your gut—anywhere but the delicate sanctuary of your mind.
We were wrong.
Today, neuroinflammation stands at the center of our understanding of almost every major brain disease. Alzheimer’s. Parkinson’s. Multiple sclerosis. Traumatic brain injury. Even depression and anxiety. They all share a common thread: the brain is on fire .
And the most terrifying part? This fire can burn for twenty years before you ever notice a single symptom.
This is the story of that fire—what ignites it, what fuels it, and what scientists are learning about how to extinguish it before it consumes everything.
The Brain’s Hidden Immune System
The Fortress Analogy
Think of your brain as a fortress—the most valuable territory your body possesses. It houses everything you are: your memories, your personality, your very consciousness. Protecting this fortress is so critical that evolution built walls unlike anywhere else in the body.
These walls are the blood-brain barrier (BBB) , a tightly sealed network of cells lining every blood vessel in your brain. Unlike the leaky capillaries elsewhere in your body, brain capillaries are sealed shut by tight junctions—like bricks packed so closely together that not even a knife blade can slip between them. Only essential nutrients like glucose and oxygen are granted passage. Everything else is turned away .
For centuries, this fortress analogy led scientists to believe the brain was entirely cut off from the immune system. They thought immune cells couldn’t enter, and therefore, inflammation couldn’t happen.
But every fortress needs guards inside the walls.
The Resident Guardians
While peripheral immune cells (like the T-cells and B-cells that patrol the rest of your body) are largely excluded from the healthy brain, the brain maintains its own dedicated defense force. These are the glial cells—specifically, microglia and astrocytes.
Microglia are the brain’s first responders. These cells make up about 10-15% of all cells in the brain, and they are constantly on patrol. Each microglia monitors its own territory, extending and retracting finger-like processes to sample the environment, checking for signs of trouble. In a healthy brain, they appear “resting”—but this is misleading. They are never truly at rest. They are always watching .
Astrocytes, named for their star-shaped appearance, are the brain’s support crew. They nourish neurons, regulate neurotransmitter levels, maintain the blood-brain barrier, and respond to injuries. When microglia sound the alarm, astrocytes rush to contain the damage .
Together, these cells form the brain’s innate immune system—a defense force capable of responding to threats without help from the rest of the body.
DID YOU KNOW?
Your brain has its own dedicated immune cells called microglia. They make up about 10-15% of all brain cells—roughly 8-12 billion microglia constantly patrolling for threats. And they’re incredibly sensitive: microglia can detect a single dying neuron from millimeters away and migrate directly to the site of injury .
When the Guards Attack
The problem is that microglia and astrocytes, designed to protect, can sometimes become the very source of destruction.
Imagine security guards who, after years of dealing with minor disturbances, become permanently jumpy. They start seeing threats everywhere. They begin using excessive force. They arrest innocent bystanders. They damage the very facility they’re supposed to protect.
This is exactly what happens in chronic neuroinflammation. The guards—microglia and astrocytes—enter a state of persistent activation. They release inflammatory chemicals meant to fight threats, but these chemicals also damage healthy neurons. They clear away debris, but sometimes they clear away living synapses. They try to contain damage, but the containment itself becomes damaging .
This is the paradox of neuroinflammation: the very system designed to save your brain can become the instrument of its destruction.
Acute vs. Chronic — Good Fire, Bad Fire
The Good Fire: Acute Neuroinflammation
Not all inflammation is bad. In fact, acute inflammation is essential for survival.
Imagine you suffer a traumatic brain injury—a fall, a car accident, a sports collision. Immediately, damaged cells release signals: “Help! We’re under attack!”
Microglia rush to the scene. They transform from their resting, ramified state into an activated, amoeba-like form capable of movement and phagocytosis—literally eating dead cells and debris. They release chemical signals that recruit more microglia and astrocytes to contain the damage .
This acute inflammatory response accomplishes several critical tasks:
- Clearance: Dead and dying cells are removed before their contents can poison surrounding tissue
- Containment: A “glial scar” forms around the injury, walling it off from healthy tissue
- Repair: Growth factors are released to promote healing and plasticity
Within days to weeks, the inflammation resolves. The microglia return to their resting state. The brain heals. This is inflammation working exactly as it should.
DID YOU KNOW?
Acute neuroinflammation is actually protective. Without it, even minor brain injuries could become catastrophic. The problem isn’t inflammation—it’s inflammation that never turns off.
The Bad Fire: Chronic Neuroinflammation
Now imagine that the fire never goes out.
Instead of resolving, the inflammatory response persists—for months, years, even decades. The microglia never return to their resting state. They remain activated, continuously pumping out inflammatory signals. The astrocytes never stop trying to contain damage that won’t go away .
This chronic neuroinflammation is fundamentally different from acute inflammation. It’s not a controlled burn; it’s a slow, smoldering wildfire that gradually consumes everything in its path.
The consequences are devastating:
Synaptic damage: Chronic inflammation attacks synapses—the connections between neurons where communication happens. Cytokines like TNF-α and IL-1β can directly impair synaptic function, weakening connections and eventually causing them to disappear. This synapse loss correlates more strongly with cognitive decline than neuron loss itself .
Neuronal death: Prolonged exposure to inflammatory chemicals pushes neurons toward apoptosis—programmed cell death. The very cells that cannot be replaced are systematically eliminated .
Blood-brain barrier breakdown: Chronic inflammation weakens the tight junctions between endothelial cells, making the blood-brain barrier leaky. This allows peripheral immune cells and toxins to enter the brain, fueling even more inflammation—a vicious cycle .
Impaired neurogenesis: The adult brain can generate new neurons in certain regions, particularly the hippocampus (memory center). Chronic inflammation suppresses this neurogenesis, reducing the brain’s ability to repair itself and adapt .
The difference between acute and chronic neuroinflammation is the difference between a controlled campfire and a wildfire that has been burning for decades. One keeps you warm. The other destroys everything.
The Cellular Players
Microglia: The Double-Edged Sword
Microglia are the stars of the neuroinflammation story—and like many stars, they have both a heroic side and a tragic flaw.
The M1/M2 Paradigm
For years, scientists classified microglial activation into two simplified states :
| State | Also Called | Function | Markers |
|---|---|---|---|
| M1 | Classical activation | Pro-inflammatory, killing threats | CD86, IL-1β, IL-6, iNOS, TNF-α |
| M2 | Alternative activation | Anti-inflammatory, repair | IL-10, Arg-1, CD206 |
In this model, M1 microglia are the aggressive, attack-mode cells. They release inflammatory cytokines, generate reactive oxygen species, and attempt to destroy whatever they perceive as a threat. M2 microglia, by contrast, are the peacekeepers. They release anti-inflammatory signals, promote tissue repair, and help restore homeostasis .
The healthy brain maintains a careful balance between these states. Acute threats trigger a temporary shift toward M1, followed by a shift toward M2 for cleanup and repair. The system returns to baseline.
In chronic neuroinflammation, this balance breaks. Microglia become locked in a prolonged M1 state. They never transition to M2. The attack never stops .
Beyond M1/M2: The New Understanding
Recent research has revealed that the M1/M2 classification is oversimplified. Microglia exist along a continuum of activation states, simultaneously expressing genes from both “types” depending on the exact signals they receive .
This complexity matters because it suggests that simply blocking all microglial activation isn’t the answer. You need to modulate their behavior—pushing them away from destructive states while preserving their protective functions.
Recent studies have identified specific molecular pathways that can shift microglial polarization. For example, FGF20 (fibroblast growth factor 20) has been shown to suppress M1-associated markers while enhancing M2-associated markers in animal models of stroke, reducing inflammation and improving outcomes .
Another promising target is NEU1 (neuraminidase 1), an enzyme that amplifies inflammatory signaling in microglia. Inhibiting NEU1 with compounds like osthole (a natural coumarin) reduces microglial activation and improves cognitive function in animal models .
DID YOU KNOW?
Microglia are incredibly dynamic cells. In their resting state, they constantly extend and retract tiny processes to survey their environment—each microglia can sample its entire territory every few hours. When they detect damage, they can transform into a fully activated, mobile state in minutes .
Astrocytes: From Support to Attack
If microglia are the brain’s front-line soldiers, astrocytes are the logistics and support crew—until they, too, join the fight.
Normal Astrocyte Functions
In a healthy brain, astrocytes perform essential tasks:
- Structural support: They physically anchor neurons and blood vessels
- Metabolic support: They supply neurons with energy substrates like lactate
- Neurotransmitter regulation: They clear excess glutamate to prevent excitotoxicity
- Blood-brain barrier maintenance: Their end-feet surround capillaries, helping maintain barrier integrity
- Synaptic regulation: They release factors that influence synapse formation and function
Reactive Astrocytes
When injury or inflammation occurs, astrocytes become “reactive.” They hypertrophy (swell), upregulate specific proteins like GFAP (glial fibrillary acidic protein), and change their behavior .
Like microglia, reactive astrocytes can take on different states:
A1 astrocytes are induced by activated microglia and become neurotoxic. They lose their normal supportive functions and begin releasing factors that can kill neurons and oligodendrocytes (the cells that make myelin).
A2 astrocytes are induced by injury and become neuroprotective. They upregulate factors that promote repair and support neuronal survival.
In chronic neuroinflammation, the balance shifts toward toxic A1 astrocytes, contributing to neuronal damage and disease progression .
The Deadly Dialogue
The real damage comes from the communication between microglia and astrocytes—a “deadly dialogue” that amplifies and perpetuates inflammation .
Activated microglia release cytokines like IL-1α, TNF-α, and C1q that can push astrocytes toward the toxic A1 state. These A1 astrocytes then release their own inflammatory signals that further activate microglia. A positive feedback loop is established: microglia activate astrocytes, astrocytes activate microglia, and the inflammation spirals .
Breaking this cycle requires targeting the communication itself—not just individual cell types.
The Signaling Pathways
The Molecular Conversation
The dialogue between cells in the inflamed brain happens through specific molecular signals. Understanding these signals reveals potential targets for intervention.
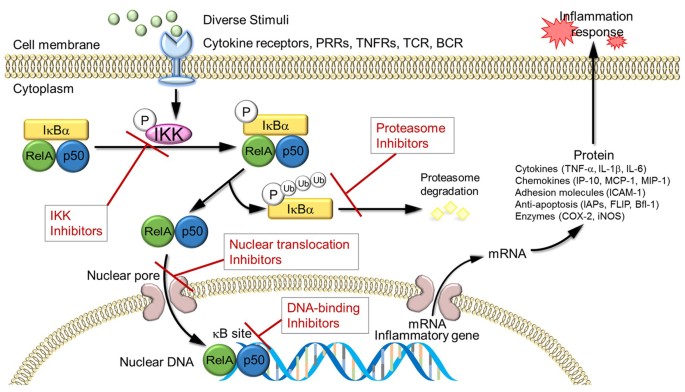
The TLR4/NF-κB Pathway: The Master Switch
Toll-like receptor 4 (TLR4) is one of the brain’s primary danger sensors. It recognizes not only bacterial components (like LPS) but also damage-associated signals released by injured cells—including amyloid-beta in Alzheimer’s disease .
When TLR4 detects a threat, it triggers a cascade that activates NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells). NF-κB acts as a “master switch” that enters the cell nucleus and turns on dozens of inflammatory genes .
The result? Production of:
- TNF-α (tumor necrosis factor-alpha)
- IL-1β (interleukin-1 beta)
- IL-6 (interleukin-6)
- COX-2 (cyclooxygenase-2)
- iNOS (inducible nitric oxide synthase)
These are the weapons microglia use to fight threats—but they also damage healthy tissue when produced chronically .
Multiple therapeutic approaches are targeting this pathway. The natural compound harmine (derived from plants) has been shown to inhibit TLR4/NF-κB signaling, reducing neuroinflammation in preclinical models . FGF20 also suppresses this pathway through mechanisms that may be independent of its receptor binding .
DID YOU KNOW?
The TLR4/NF-κB pathway is so central to inflammation that it’s been conserved across hundreds of millions of years of evolution. Essentially the same signaling system exists in fruit flies and humans—a testament to its fundamental importance .
The NLRP3 Inflammasome: The Amplifier
If TLR4/NF-κB is the switch that turns on inflammation, the NLRP3 inflammasome is the amplifier that makes it truly destructive.
The NLRP3 inflammasome is a multi-protein complex that detects cellular stress and responds by activating caspase-1, which then cleaves pro-IL-1β into its active, secreted form. This massively amplifies the inflammatory response .
Why does this matter? Because IL-1β is one of the most potent inflammatory cytokines in the brain. It can directly damage neurons, disrupt synaptic function, and recruit more immune cells to the site of inflammation.
The NLRP3 inflammasome has been implicated in virtually every neurodegenerative disease. In Alzheimer’s, amyloid-beta can directly activate it. In Parkinson’s, alpha-synuclein does the same. In traumatic brain injury, cellular debris triggers its activation .
Compounds that inhibit NLRP3 are being actively developed as potential therapeutics. Harmine and its derivatives have shown promise in preclinical models .
The TREM2 Pathway: The Brake
Every inflammatory system needs brakes—ways to dampen the response once the threat is contained. TREM2 (Triggering Receptor Expressed on Myeloid cells 2) is one of the most important brakes in the brain.
TREM2 is a receptor on microglia that recognizes lipids and other damage-associated signals. When activated, it promotes microglial survival, phagocytosis, and a protective, repair-oriented phenotype. It essentially tells microglia: “Clear the debris, but don’t attack healthy tissue” .
Genetic variants in TREM2 significantly increase Alzheimer’s risk—by about 3-4 times for some mutations. This tells us that when the brake fails, the inflammatory fire burns hotter .
Recent research has shown that FGF20 can upregulate TREM2 expression, shifting microglia toward a protective state. This may be one mechanism by which it reduces neuroinflammation in stroke models .
The MAPK Pathway: The Intensifier
MAPK (mitogen-activated protein kinase) pathways are signaling cascades that amplify and diversify inflammatory signals. They act downstream of TLR4 and other receptors, leading to the production of multiple inflammatory mediators .
Different MAPK pathways (p38, JNK, ERK) have distinct but overlapping roles in neuroinflammation. Inhibiting these pathways is another therapeutic strategy being explored.
The Interplay: Why Combination Therapy Matters
These pathways don’t operate in isolation. They form a complex network with extensive crosstalk :
- TLR4/NF-κB activation increases NLRP3 inflammasome components
- NLRP3 produces IL-1β, which signals through its own receptor to amplify NF-κB
- TREM2 can inhibit both TLR4 and NLRP3 signaling
- MAPK pathways intersect with all of the above
This complexity explains why single-target drugs have often failed in clinical trials. You might block one pathway, but the others can compensate. The future of neuroinflammation treatment likely lies in combination approaches that target multiple nodes simultaneously .
The Blood-Brain Barrier—Gatekeeper Under Siege
The Fortress Walls
The blood-brain barrier (BBB) is not a single structure but a system:
- Endothelial cells form the walls of brain capillaries, connected by tight junctions
- Pericytes wrap around endothelial cells, providing structural support and regulating permeability
- Astrocyte end-feet surround the vessels, completing the barrier
- Basement membrane provides an additional layer of physical separation
This system is so effective that it blocks over 98% of small molecules and virtually all large molecules from entering the brain. It’s why treating brain diseases is so difficult—most drugs simply can’t get through.
When the Barrier Breaks
Chronic inflammation attacks the BBB at multiple levels :
Tight junction disruption: Inflammatory cytokines like TNF-α and IL-1β signal endothelial cells to loosen their tight junctions. The bricks develop gaps. The wall becomes porous.
Endothelial activation: Inflammatory signals cause endothelial cells to express adhesion molecules (like ICAM-1 and VCAM-1) on their surface. These act as “docking stations” for peripheral immune cells.
Immune cell infiltration: Activated T-cells, B-cells, and monocytes can now stick to the vessel wall and squeeze through the gaps. Once inside, they bring peripheral inflammatory mechanisms directly into the brain.
Matrix metalloproteinase activation: These enzymes degrade the basement membrane, further compromising barrier integrity.
The result is a vicious cycle: inflammation weakens the BBB, allowing peripheral immune cells and inflammatory molecules to enter, which fuels more inflammation, which further weakens the BBB .
DID YOU KNOW?
The blood-brain barrier is so selective that even water doesn’t simply diffuse through—it must pass through specialized channels called aquaporins. This tight control is why brain swelling (cerebral edema) is so dangerous; fluid can enter faster than it can leave .
Therapeutic Implications
The BBB presents both a challenge and an opportunity for treating neuroinflammation.
The challenge: Any drug targeting neuroinflammation must be designed to cross the BBB. Many promising compounds fail in clinical trials simply because they can’t reach therapeutic concentrations in the brain .
The opportunity: The BBB itself can be targeted. Drugs that strengthen tight junctions or inhibit matrix metalloproteinases could help preserve barrier integrity, breaking the vicious cycle of inflammation and leakage.
Recent advances include:
- Nanoparticle carriers that can ferry drugs across the BBB
- Trojan horse strategies that disguise drugs as molecules the BBB normally transports (like glucose or transferrin)
- Focused ultrasound that temporarily opens the BBB at specific locations
Neuroinflammation Across Diseases
The Common Thread
One of the most important insights of the past decade is that neuroinflammation is not specific to any single disease. Instead, it’s a common thread running through almost all neurological conditions—each with its own unique triggers, but sharing the same fundamental inflammatory mechanisms .
Alzheimer’s Disease
In Alzheimer’s, the trigger is protein aggregation—specifically, amyloid-beta plaques and tau tangles.
Amyloid-beta is directly detected by microglial TLR4 and other receptors, triggering an inflammatory response. Initially, this is protective: microglia attempt to clear the plaques. But over years of chronic exposure, they become exhausted and dysfunctional. They shift toward a pro-inflammatory state, releasing cytokines that damage synapses and neurons .
Genetic risk factors tell the same story:
- APOE4, the strongest genetic risk factor for late-onset Alzheimer’s, impairs microglial function and lipid metabolism
- TREM2 variants impair microglial response to damage
- CD33 variants affect microglial activation
The inflammation in Alzheimer’s isn’t secondary—it’s central to the disease process. PET imaging studies show that microglial activation correlates with cognitive decline even more strongly than amyloid burden .
Recent therapeutic developments:
- cPLA2 inhibitors: USC researchers have identified compounds that inhibit calcium-dependent phospholipase A2 (cPLA2), an enzyme linked to inflammation in APOE4 carriers. These compounds cross the blood-brain barrier and reduce neuroinflammation in preclinical models .
- Harmine: This natural compound inhibits multiple inflammatory pathways (TLR4/NF-κB, NLRP3) while also reducing tau hyperphosphorylation .
- NEU1 inhibitors: Targeting neuraminidase 1 reduces microglial activation and improves cognition in animal models .
DID YOU KNOW?
People with certain autoimmune diseases (like rheumatoid arthritis and psoriasis) have a higher risk of developing Alzheimer’s. This suggests that systemic inflammation can cross into the brain and accelerate neurodegeneration—another example of the brain-body connection .
Parkinson’s Disease
In Parkinson’s, the trigger is alpha-synuclein aggregation. Misfolded alpha-synuclein forms Lewy bodies inside neurons, and when neurons die, they release alpha-synuclein into the surrounding tissue.
Microglia detect extracellular alpha-synuclein through TLR4 and other receptors, triggering inflammation. This inflammation then kills more neurons, releasing more alpha-synuclein—another vicious cycle .
The inflammation in Parkinson’s is most intense in the substantia nigra, the brain region where dopamine neurons die. PET imaging shows activated microglia in this region even in early-stage disease.
Multiple Sclerosis
Multiple sclerosis is fundamentally an autoimmune disease, but inflammation is the mechanism of damage.
In MS, autoreactive T-cells and B-cells cross the blood-brain barrier and attack myelin—the insulating sheath around axons. This triggers a massive inflammatory response involving both peripheral immune cells and resident microglia/astrocytes .
The inflammation in MS is more acute and relapsing-remitting than in Alzheimer’s or Parkinson’s, but chronic progressive MS involves the same smoldering neuroinflammation seen in other diseases .
Recent advances:
- BTK inhibitors: Bruton’s tyrosine kinase inhibitors target microglial activation and are showing promise in progressive MS .
- B-cell depletion therapies (like ocrelizumab) reduce inflammation by eliminating the B-cells that drive autoimmunity .
Traumatic Brain Injury and Beyond
Even acute injuries can trigger chronic neuroinflammation. Following traumatic brain injury, microglial activation can persist for years—long after the initial injury has healed. This chronic inflammation may explain why TBI increases risk for later developing Alzheimer’s and other neurodegenerative diseases .
Other conditions with a neuroinflammatory component include:
- ALS (amyotrophic lateral sclerosis)
- Frontotemporal dementia
- Stroke
- Depression and anxiety
- Schizophrenia
- Autism spectrum disorders
The Therapeutic Frontier
Learning from Past Failures
The history of anti-inflammatory treatments for brain diseases is littered with failed clinical trials. NSAIDs (non-steroidal anti-inflammatory drugs) like ibuprofen and naproxen looked promising in observational studies but failed to prevent or treat Alzheimer’s in randomized trials .
Why did they fail? Several reasons:
Timing: The trials may have intervened too late. By the time symptoms appear, neuroinflammation has been burning for years or decades. You can’t put out a decades-old wildfire with a garden hose.
Targeting: NSAIDs are broad-spectrum and relatively weak. They may not adequately penetrate the brain or sufficiently suppress the specific inflammatory pathways driving neurodegeneration.
Complexity: Inflammation is complex and context-dependent. Simply blocking all inflammation might also block protective inflammatory responses.
New Approaches: Targeted Modulation
The new generation of therapies aims for modulation rather than suppression—pushing the inflammatory response back toward homeostasis rather than eliminating it entirely.
Microglial Polarization Modulators
Several compounds are being developed to shift microglia from harmful M1 states toward protective M2 states:
Inflammasome Inhibitors
Targeting the NLRP3 inflammasome is a major focus of drug development. Several compounds are in various stages of testing, including:
- MCC950 (small molecule inhibitor)
- DFV890 (clinical trials for various inflammatory conditions)
- Harmine derivatives (preclinical)
cPLA2 Inhibitors
USC researchers have developed selective cPLA2 inhibitors that cross the blood-brain barrier and reduce neuroinflammation in APOE4-related models. These compounds are designed to inhibit pathological enzyme activity while preserving normal cellular function .
BTK Inhibitors
Bruton’s tyrosine kinase inhibitors target microglial activation and are being tested in progressive multiple sclerosis, with promising early results .
The Promise of CAR-T and Cell-Based Therapies
Perhaps the most exciting frontier is the application of advanced immunotherapies to neuroinflammation. Just as CAR-T cells have revolutionized cancer treatment, similar approaches might one day be used to modulate brain inflammation .
The concept: engineer immune cells to specifically target and suppress pathogenic microglia or autoreactive lymphocytes, while leaving protective responses intact. This is early-stage research, but the potential is enormous.
Lifestyle Interventions
While drug development proceeds, evidence continues to accumulate that lifestyle factors can modulate neuroinflammation:
Diet: The Mediterranean diet (rich in omega-3 fatty acids, polyphenols, and fiber) has anti-inflammatory effects throughout the body, including the brain. Omega-3s in particular can shift microglia toward protective states .
Exercise: Physical activity reduces systemic inflammation and promotes the release of factors that support brain health. Animal studies show exercise can shift microglial polarization toward M2 states.
Sleep: During sleep, the glymphatic system clears waste products from the brain, including inflammatory mediators. Chronic sleep deprivation increases neuroinflammation.
Stress reduction: Chronic stress elevates cortisol and other stress hormones that can promote inflammation. Mindfulness and meditation have been shown to reduce inflammatory markers.
The Future—Putting Out the Fire
Biomarkers: Detecting the Fire Early
One of the biggest challenges in treating neuroinflammation is detecting it before it causes irreversible damage. We need ways to measure the fire while it’s still smoldering, not after it’s already consumed critical tissue.
Promising biomarkers include :
Imaging biomarkers:
- PET tracers for TSPO (translocator protein), which is upregulated on activated microglia
- PET tracers for other inflammatory targets (under development)
Fluid biomarkers:
- GFAP (glial fibrillary acidic protein) in blood or CSF—reflects astrocyte activation
- YKL-40 (also called CHI3L1)—reflects microglial/astrocyte activation
- sTREM2 in CSF—reflects microglial response
- Inflammatory cytokines (TNF-α, IL-6, IL-1β)—less specific but informative in context
Emerging biomarkers:
- NFL (neurofilament light chain)—reflects axonal damage, not inflammation specifically, but correlates with disease activity
- NEU1 activity—potential new marker under investigation
The goal is a panel of biomarkers that can detect neuroinflammation early, track its progression, and monitor response to therapy.
Precision Medicine: Not All Fires Are the Same
Just as fires have different causes and behaviors, neuroinflammation differs between individuals and diseases. The future of treatment lies in matching the intervention to the specific inflammatory profile .
By genotype:
- APOE4 carriers may benefit from different anti-inflammatory strategies than non-carriers
- TREM2 variant carriers might need therapies that enhance TREM2 function
- Individuals with specific inflammatory gene variants might respond differently to particular drugs
By disease stage:
- Early inflammation may be primarily microglial and responsive to modulation
- Late-stage inflammation involves astrocytes, BBB breakdown, and peripheral immune infiltration—requiring different approaches
By trigger:
- Protein-aggregation-driven inflammation (Alzheimer’s, Parkinson’s) might need combined anti-aggregation and anti-inflammatory approaches
- Autoimmune-driven inflammation (MS) might need immune-modulating therapies
- Trauma-induced inflammation might need acute anti-inflammatory intervention followed by long-term monitoring
Combination Therapy: The Cancer Model
Oncologists learned years ago that single drugs rarely cure cancer. The future belongs to combinations :
Anti-inflammatory + anti-aggregation: In Alzheimer’s, combining anti-amyloid antibodies with anti-inflammatory drugs might be more effective than either alone.
Multiple pathway targeting: Hitting TLR4/NF-κB, NLRP3, and MAPK simultaneously might be more effective than targeting any single pathway.
Central + peripheral: Targeting both brain-resident inflammation (microglia) and peripheral immune infiltration might break the vicious cycle.
The Ultimate Goal: Prevention
The holy grail is preventing neuroinflammation before it starts—or extinguishing it so early that neurons never suffer damage.
This means:
- Identifying at-risk individuals early (through genetics, biomarkers, lifestyle factors)
- Intervening in midlife, decades before symptom onset
- Using safe, long-term interventions (lifestyle, nutraceuticals, low-dose pharmaceuticals) to keep the inflammatory fire from ever igniting
The Fire Within
Neuroinflammation is not a disease. It is a process—a mechanism—that underlies almost everything that can go wrong in the human brain. It is the final common pathway through which diverse triggers (proteins, genes, injuries, infections) ultimately destroy the tissue that makes us who we are.
But understanding this process gives us power. Power to detect the fire early. Power to modulate it without extinguishing it entirely. Power to protect the brain while preserving the protective functions of inflammation.
The fire within is real. But for the first time in human history, we are learning how to control it.
Continue Exploring
This article is part of Utopedia’s Brain Health & Neuroscience series. To deepen your understanding, explore these related entries:
Alzheimer’s Disease: The Gradual Fading of the Mind’s Tapestry
The complete guide to plaques, tangles, and the 20-year silent phase—now available in the Utopedia library.
The Blood-Brain Barrier: Fortress Under Siege
How the brain protects itself, and what happens when the walls crumble.
Microglia: The Brain’s Guardians
Deep dive into the cells that protect and sometimes destroy our most precious organ.
Astrocytes: The Forgotten Half of Brain Health
Beyond neurons: the star-shaped cells that support everything we think and do.
Inflammation Across the Body: The Brain Connection
How gut health, joint pain, and heart disease connect to what happens inside your skull.
APOE: The Gene That Shapes Brain Aging
Everything you need to know about the most important genetic risk factor for Alzheimer’s.
Clinical Trials in Neuroinflammation: 2026 Update
What’s in the pipeline, what failed, and what’s showing promise.
Join the UtopiaCircle Campus Network
This article is just the beginning. At UtopiaCircle, we’re building a community of curious minds exploring the frontiers of science, medicine, and human potential.
When you join our Campus Network, you get:
- Early access to new Utopedia entries before public release
- Monthly research summaries from leading journals
- Live Q&A sessions with researchers and clinicians
- Discussion forums to explore ideas with fellow learners
- Opportunities to suggest topics for future articles
Leave a comment below: What aspect of neuroinflammation most intrigues or concerns you? Have you or a loved one been affected by a condition involving brain inflammation? Your stories and questions shape the content we create.
Share this article with someone who needs to understand the hidden fire within.
Together, we move from curiosity to understanding.
References
- Zhu L, et al. FGF20 alleviates neuroinflammation in ischemic stroke by modulating microglial polarization via TREM2-TLR4/NF-κB pathway. Cell Immunol. 2026;423-424:105095.
- Shi FD, Wee Yong V. Neuroinflammation Across Neurological Diseases. Science. 2025;388(6753).
- Shen X, et al. Harmine and its derivatives: A promising multi-target therapeutic avenue for Alzheimer’s disease. Neuroscience. 2026. Online ahead of print.
- Müller L, et al. Neuroimmune crosstalk in chronic neuroinflammation: microglial interactions and immune modulation. Front Cell Neurosci. 2025;19:1575022.
- The role of inflammation in neurological disorders: a brief overview of multiple sclerosis, Alzheimer’s, and Parkinson’s disease. Front Neurol. 2024;15:1439125.
- Alzarea SI, et al. A network pharmacology and in silico approach to target NEU1-mediated microglial activation in neuroinflammation: Validation in an LPS-induced mouse model. Comput Biol Med. 2025;178:109652.
- Microglial dysfunction in Alzheimer’s disease: Mechanisms, emerging therapies, and future directions. Exp Neurol. 2025;392:115374.
- Masrori P, et al. The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD. Mol Neurodegener. 2022;17(1):22.
- Sadybekov AV, et al. Development of potent, selective cPLA2 inhibitors for targeting neuroinflammation in Alzheimer’s disease and other neurodegenerative disorders. npj Drug Discovery. 2026.
- Molecular insights into glial neuroimmune cross reactivity with CNS antigens and its role in neuroinflammation. Inflammopharmacology. 2026.